Transcription Readthrough in Non-Pathological Conditions
Transcription termination is vital for producing functional mRNAs and proteins. Transcription Readthrough (TRT) occurs when the transcription machinery fails to read stop signals, resulting in longer aberrant transcripts that may disrupt cellular function. This phenomenon is linked to stress responses, viral infections, and cancer. TRT transcripts may lead to the invasion of downstream neighbours and even form mRNA molecules that combine coding elements from different genes. However, their prevalence and functional impact in healthy conditions have remained largely unexplored.
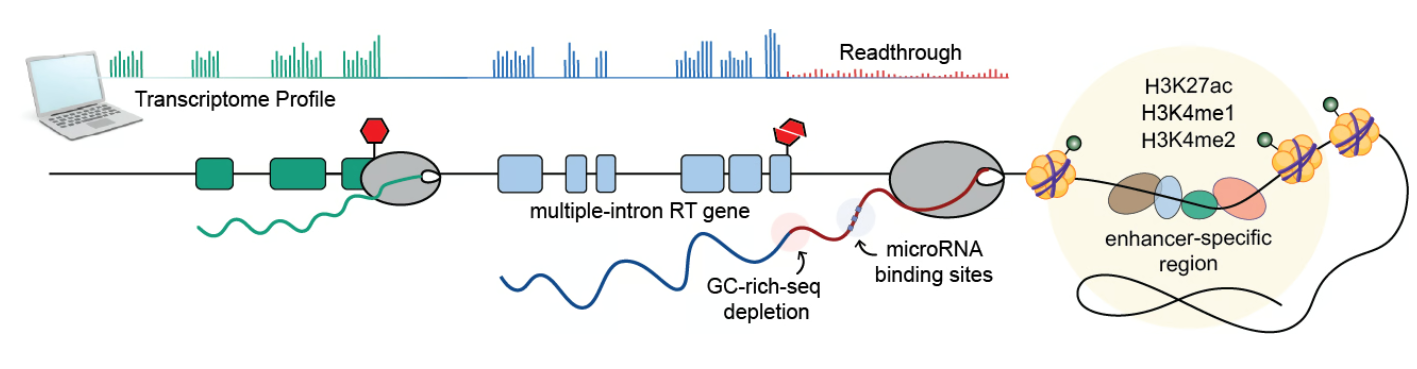
In this study, we aimed to uncover the prevalence of readthrough transcripts in healthy human tissues and elucidate the factors influencing their production. For this, we analyzed nearly 3000 transcriptome profiles from the Genotype-Tissue Expression (GTEx) project using established bioinformatics tools. We inspected high read coverage downstream of all expressed genes in these samples to infer readthrough.
We found that at least 30% of all protein-coding genes produce RT transcripts, regardless of the transcriptional levels of their host genes, consistent with previous studies. Only a small fraction (<1% across tissues) reaches downstream genes, contrasting with cancer-associated TRT events. This suggests that read-in events may be exclusive to extreme cases of cellular stress. Additionally, we observed a negative correlation between transcription readthrough and nearby gene activity, supporting the role of RNA polymerase collisions in preventing readthrough.
We discovered a striking correlation between transcription readthrough and the number of introns in expressed genes. Genes lacking introns are less prone to readthrough (<5%), while over 30% of genes with 20+ introns exhibit TRT. This suggests that splicing efficiency may affect molecular processes at gene ends, consistent with previous findings showing that unspliced mRNAs often produce readthrough transcripts.
Our analysis also revealed a significant scarcity of GC-rich sequences at the 3’ end of RT genes. This depletion of GC clusters is correlated with a shorter window for RNA cleavage, which may increase the likelihood of readthrough. Interestingly, we did not observe the previously described depletion of polyA signals in stress-induced transcription readthrough, indicating that their presence does not determine TRT occurrence, at least in healthy tissues.
We also analyzed the chromatin landscape of readthrough transcribed regions using molecular profiles from the Epigenome Roadmap Project. Our findings show that the regions surrounding the termination site of RT genes are associated with accessible regulatory chromatin, consistent with previous observations under stress or HIV infection. Moreover, regions surrounding RT tails exhibit higher levels of enhancer-specific histones, suggesting a role for enhancer activity in restricting abnormal elongation. However, it is unclear whether these chromatin alterations are the cause or consequence of readthrough transcription.
Our enrichment analysis suggested a potential role for RT transcripts in normal conditions, prompting us to hypothesize that transcription readthrough may interfere with protein production via miRNA-mediated gene regulation by extending miRNA binding regions. To test this hypothesis, we identified putative miRNA binding sites along the tail of these transcripts and respective genes. We found that more than 20% of all RT genes potentially act as sponges - capturing specific miRNAs through multiple binding sites - for a large proportion of miRNAs across different tissues.
This study sheds light on the prevalence, regulation, and potential functional impact of transcription readthrough in healthy human tissues, opening avenues to further explore its intricate molecular mechanisms and physiological significance.
See full publication in Communications Biology and Behind the Paper in Springer Nature Research Communities.